Introduction
In recent years, ammonia borane (AB) and its derivatives have gained momentum as promising hydrogen storage materials. Hydrogen is mainly generated from ABs using thermolysis and hydrolysis. However, only hydrolysis of AB has been extensively studied because thermolysis reported various disadvantages. Hydrolysis of AB requires a suitable catalyst to accelerate the reaction and generate hydrogen at reasonable rates. To date various metal and some non-metal catalysts have been used to enhance the hydrogen elimination of AB. In this review we report the use of various catalysts for the dehydrogenation of AB and its derivatives to generate hydrogen efficiently. Work is ongoing to find efficient catalysts for hydrogen release from AB materials which it is hoped will lead to use of these compounds as chemical hydrogen storage systems for on-board vehicular applications, replacing fossil fuels and boosting the hydrogen economy.
Ammonia borane as a chemical hydrogen storage system
AB has a tetragonal structure and is a material with potential for chemical hydrogen storage. AB has a high hydrogen content (19.6 wt%) and is soluble in water and relatively polar solvents like methanol.1,2 AB was first synthesised for use in rockets and jets in the mid-1950s as a high-energy fuel. AB is usually now prepared on a small scale by reaction between sodium borohydride (NaBH4) and ammonium formate (HCO2NH4) in the presence of dioxane with high yield (≥ 95%) and high purity (≥ 98%). Usefully, AB is nontoxic and highly stable under ambient conditions, thus facilitating its application as an efficient hydrogen storage material.1,3
Hydrogen stored in the AB adduct can be released on demand using thermal decomposition or solvolysis (hydrolysis or methanolysis). Thermal dehydrogenation of AB is a four-step process and can eliminate three equivalents of hydrogen. The first step is melting of AB (Eq. 1). AB starts to dehydrogenate at approximately 100℃ in the second step and release 6.5 wt% of H2 (Eq. 2). The second equivalent of H2 is evolved in the third step which occurs at around 150℃ (Eq. 3). The final step, which requires elevated temperatures (> 1200℃), releases the third equivalent of H2 (Eq. 4).
Thermolysis of AB has some disadvantages; it requires a long induction time (∼3h), high temperatures and unwanted gaseous by-products like borazine (B3N3H6) and ammonia (NH3) are liberated during the reactions.4 Various approaches including catalysis5,6 have been considered to overcome these issues.
NH3BH3 (g) → NH3BH3 (l) (Eq. 1)
NH3BH3 (l) → 1/n [NH2-BH2]n (s) + H2 (g) (Eq. 2)
1/n [NH2-BH2]n (s) → 1/n [NH-BH]n (s) + H2 (g) (Eq. 3)
1/n [NH-BH]n (s) → N-B (s) + H2 (g) (Eq. 4)
According to the kinetic study of the linear AB by Nguyen et al.7 the H2 elimination reaction of AB has a higher reaction barrier than the dissociation energy of the complex. It is noted that the reaction energy barrier for the dehydrogenation of AB is 43.5 kJ/mol above the B-N bond dissociation energy to generate BH3 and NH3. As a result, dissociation of the B-N bond is predicted to be favoured over evolution of H2. Consequently, dehydrogenation barriers need to be reduced below the B-N dissociation limit with suitable catalysts to enable evolution of H2 from these systems.
An alternative method to form hydrogen from AB for mobile applications at ambient temperature is solvolysis. Hydrolysis of 1 mol AB (Eq. 5) can release 3 mol of H2.
NH3BH3 (aq) + 2H2O (l) → NH4+ (aq) + BO2- (aq) + 3H2 (g) (Eq. 5)
The hydrolytic dehydrogenation of AB is monitored by 11B NMR where a quartet around δ = -24.0 ppm is changed to a singlet around δ = 10.0 ppm when AB converts into borate (BO2-).8 The hydrolytic dehydrogenation of AB does not occur at an appreciable rate at room temperature without a suitable catalyst since AB is relatively stable to hydrolysis in aqueous solution. Fig. 1 shows the mechanism, catalysts and limitations of catalysed ammonia borane hydrolysis.
Ammonia borane derivatives as chemical hydrogen storage systems
Alternatives to AB such as methylamine borane,6 sec-butylamine borane,10 hydrazine borane11 and ethylene diamine bisborane12,13 have been investigated because the nitrogen substituted derivatives of borazine and substituted amines are less volatile than borazine and ammonia (byproducts of the decomposition of AB).
Hydrazine borane (HB) has been considered as an efficient hydrogen storage candidate due to its high hydrogen content (15.4 wt% H) and higher stability in the solid form at room temperature.14,15 HB can be easily synthesised from a reaction of dihydrazine sulfate with sodium borohydride in dioxane at room temperature16,17 (Eq. 6). Hydrogen can be generated from HB using various methods such as hydrolysis and decomposition,18,19 methanolysis20 and solid state thermolysis.11 HB has been shown to give off up to 6.5 wt% hydrogen very slowly upon heating to 150℃ for 16h.11 However, catalysts can be used to maximise the efficiency of HB as a promising hydrogen storage material.
Hydrazine bisborane (HBB), another derivative of AB which contains 16.8 wt% of theoretical hydrogen capacity can be easily prepared by mixing hydrazine sulfate with sodium borohydride in dioxane11 (Eq. 7). The decomposition of HBB resulted in more than 13.0 wt% of hydrogen at around 100℃ with high purity (> 99 wt%).21 However, the application of HBB as a potential hydrogen storage material in the automotive industry is limited by both the slow dehydrogenation rate, especially at low temperatures, and evolution of poisonous ammonia during decomposition. Improvement in the dehydrogenation properties of HBB has been achieved with the introduction of catalysts.
(N2H5)2SO4 + 2NaBH4 → 2N2H4-BH3 + Na2SO4 + 2H2 (Eq. 6)
N2H6SO4 + 2NaBH4 → N2H4(BH3)2 + Na2SO4 + 2H2 (Eq. 7)
Cyclic amine boranes as chemical hydrogen storage systems
Cyclic amine boranes (CABs) are nitrogen containing donor-acceptor complexes with the general formula CnH2n+1N.BX3 (n = 2-7, X = H, CH3, F, Cl, Br, I). They are formed by the coordination of the respective cyclic amines and a borane group via the formation of a dative B-N bond. One considered application of these compounds is as potential chemical hydrogen storage materials for onboard vehicular applications.22–25 The three-membered ring aziridine borane was initially synthesised with high level quantum chemical calculations revealing the hydrogen elimination reaction to be slightly endothermic.
The free energy profile for the release of H2 from aziridine borane shows that the calculated energy for the transition state is high (163.2 kJ/mol); therefore, dissociation is the favourable process.22 Thermochemical and kinetic studies of azetidine borane also predicted that the uncatalysed dehydrogenation pathway has a higher activation barrier than the calculated B-N bond dissociation energy, hence favouring dissociation of the complex over hydrogen elimination.26 The thermodynamic properties for five CABs [CnH2n+1N.BH3 (n = 2-6)]: aziridine (n = 2), azetidine (n = 3), pyrrolidine (n = 4), piperidine (n = 5) and azepane (n = 6) boranes and their associated dehydrogenated products were investigated theoretically. The dehydrogenation reaction of aziridine borane was found to be endothermic while for the other four boranes it was found to be exothermic. The reaction barrier for the dehydrogenation of aziridine, azetidine, pyrrolidine, piperidine and azepane boranes at 298.15 K is 177.0, 172.0, 166.5, 170.3 and 167.4 kJ/mol respectively. The B-N bond dissociation energies for all five CAB adducts at the same temperature were found to be less than the activation barriers for the evolution of H2. Therefore, it was again predicted that the B-N bond breaks before H2 is eliminated.24
Although the dehydrogenation of both linear and cyclic ABs showed a higher barrier than the dissociation energy of the complex, hydrogen elimination is favoured when a BH3 catalyst is used. The BH3 catalyst can reduce the reaction barrier and facilitate H2 evolution from AB and CAB and is discussed later in this review.
Catalysts and their roles in enhanced dehydrogenation of linear AB
Catalysts provide a selective route to the desired product by lowering the activation barriers and speeding up chemical reactions. In the dehydrogenation of AB, catalysts play an important role in lowering the decomposition temperature, improving the dehydrogenation rate and suppressing the impurities in hydrogen materials. The impact of the various catalysts during thermolysis as well as hydrolysis of AB to generate H2 is discussed in this section.
Thermolysis of AB with catalysts
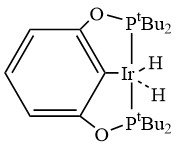
To facilitate fast hydrogen production from AB at low temperatures, an effective catalyst is required in developing potential on-board hydrogen storage systems. The rapid dehydrogenation of AB in tetrahydrofuran occurred with an iridium pincer complex catalyst (Fig. 2) at room temperature resulting the formation of one equivalent of H2 along with a dehydrogenated product [NH2BH2]5. This Ir-catalysed dehydrogenation was completed within 14 min at room temperature. A dormant form of the catalyst (metal borohydride complex) is produced over time from a reaction of BH3 with a hydride on Ir but the active form of the catalyst can be efficiently regenerated in the presence of H2. Hydrogen release of AB with the Ir pincer system showed impressive rates and potent catalyst regeneration representing a promising initial step towards the development of AB as a feasible on-board hydrogen storage system.27
Hydrogen elimination of AB was shown to be catalysed by a Ru-complex where up to 1 equivalent of H2 is evolved from AB with only 0.03 mol% Ru within 5 min at room temperature. Catalysis of dehydrogenation of AB with the Ru-complex offers a new opportunity for approaching desired hydrogen storage goals because of the mild reaction condition, low catalyst loading and prompt release of H2.28
In the presence of cationic Pd (II) complexes, dehydrogenation of AB occurs in the most efficient way to generate 2.0 equivalent of H2 at 25℃ in less than 60 s.29
Hydrolysis of AB with catalysts
The catalytic hydrolysis of AB has been extensively studied to provide reasonable rates of hydrogen formation and accelerate the hydrolysis reaction. Aqueous AB can rapidly release hydrogen with an acid30 or metal catalyst31 at room temperature. Fig. 3 represents the mechanism of AB hydrolysis in the presence of catalyst. An activated complex is formed during hydrolysis when AB interacts with the active site of the catalyst. This complex then reacts with water to produce H2.32
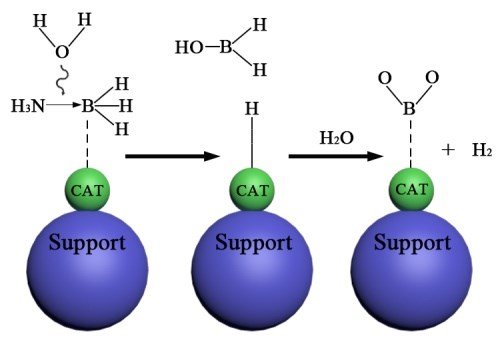
Various efforts have been used to generate catalysts with high efficiency for AB hydrolysis. To improve the catalyst specific activity, producing catalysts with a large surface area and sufficient surface-active sites is important. Therefore, metal nanoparticles or nanoclusters with higher surface area and smaller particle size have been widely reported.34–37 However, the issue of particle agglomeration remains a challenge and can be mitigated by using suitable stabilising agents.38 To stabilise transition metal nanoparticles, the prepared nanoparticles have been supported on metal oxides or oxide surfaces (TiO2, CeO2, SiO2, Al2O3), porous materials (metal-organic frameworks or zeolites) or carbonaceous materials (carbon, graphene or carbon nanotubes). Use of these materials can lead to the formation of metal nanoparticles with tunable porosities and size distribution. However, selecting a suitable stabilising agent for a particular reaction is critical.
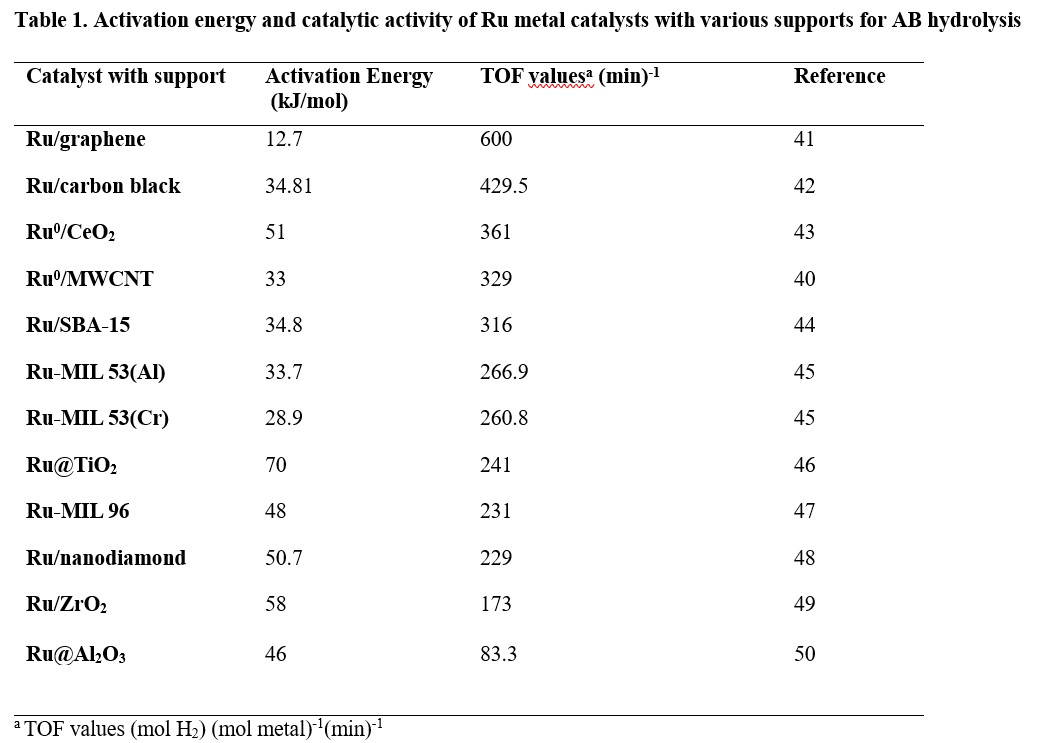
There are several requirements for an ideal supporting agent to obtain a high catalytic activity such as well-defined chemical composition, surface chemistry and pore size distribution, high purity and easy separation from the reaction medium.39 The turnover frequencies and activation energies of previously reported Ru catalysts with various supporting agents used in AB hydrolysis is shown in Table 1 with carbon-based catalysts like Ru/graphene and Ru/MWCNT (Ru nanoparticles supported on multiwalled carbon nanotubes) showing outstanding catalytic activity for hydrolysis of AB. The high dispersion of nanoparticles and elevated interaction effect of carbon nanotubes with Ru metal40 results in carbon-based materials being attractive catalyst supports compared to other stabilising materials for Ru-metal catalysed AB hydrolysis.
Both noble and non-noble metals are commonly used as catalysts in ammonia borane hydrolysis and are discussed in detail below.
Noble metal-based catalysts in hydrolysis of AB
Pt and Pt-based alloys show vigorous release of H2 during the hydrolysis of AB compared to the other noble metal-based catalysts such as Rh, Pd and Au, with the evolved H2 to AB ratio up to 3.0. The catalytic activities are ordered 20 wt% Pt/C > 40 wt% Pt/C > PtO2 > Pt black > K2PtCl4 (Fig. 4). The carbon-supported Pt catalysts display excellent performance among the other Pt-based catalysts, especially 20 wt% Pt/C which shows very high catalytic activity and reaction completion in less than 2 minutes.8 Unfortunately, these noble metal-based catalysts cannot be widely used due to the high cost and scarcity of noble metals. Thus, the development of other efficient, low cost and stable catalysts with high catalytic performance is of great interest.
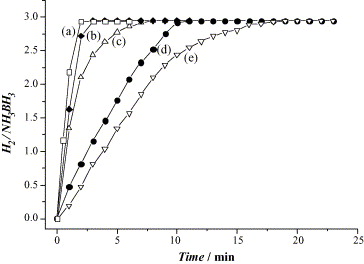
Fig. 4. Hydrogen release from AB solution in the presence of (a) 20 wt% Pt/C, (b) 40 wt% Pt/C, (c) PtO2, (d) Pt black and (e) K2PtCl4.8 Reproduced with permission.
Non-noble metal catalysts in hydrolysis of AB
Non-noble metal-based catalysts such as Co, Ni, Fe and Cu are found to have good catalytic activity for the hydrolysis of AB. Among them Co-based materials have previously shown excellent catalytic performance.51–53 Amorphous and well-dispersed Co nanoparticles without any dispersing agent can be prepared in situ at room temperature and show high catalytic performance (1116 Lmol-1min-1) and excellent recycling activity for the release of hydrogen from aqueous AB under ambient conditions.54
In order to improve the gravimetric hydrogen storage capacity (GHSC), CoCl2-catalysed hydrolysis of the AB-H2O system has been investigated by systematically changing the stoichiometric amount of water. An effective GHSC of 7.8 wt% has been reached at 25℃ with a higher hydrogen generation rate of 21 ml(H2).min-1.55 Sun and co-workers56 combined Co and Pd in variable proportions synthesised by the reduction of cobalt acetylacetonate and palladium bromide in the presence of oleylamine and trioctylphosphine. Among all the catalysts with various proportions of Co/Pd they investigated, Co35Pd65 nanoparticles exhibited the highest catalytic activity and durability. The activation energy for the hydrolysis reaction is 27.5 kJ/mol with a reaction completion time of 5.5 min which is comparable to the best Pt-based catalysts reported.56
Ni-based catalysts have been the focus of recent studies as they are cheaper and more environmentally friendly compared to Co; compared to other non-noble metal catalysts like Co, Ni is considered to be a relatively more environmentally friendly catalyst because the by-products of AB hydrolysis using Ni catalysts are considered to be environmentally benign. In addition, Ni-based catalysts can be easily separated from the reaction solution. Zhou et al.57 prepared three types of Ni nanoparticles supported on carbon (Ni/C) using a Ni-MOF precursor by (a) reduction with KBH4, (b) calcination at 700℃ under Ar, and (c) a combination of calcination and reduction, the products being labelled as Ni/C-1, Ni/C-2 and Ni/C-3. The specific surface areas of Ni-MOF, Ni/C-1, Ni/C-2 and Ni/C-3 are 1239, 33, 470 and 451m2g-1 respectively. It was found that catalytic hydrolysis of AB in the presence of Ni/C-3 shows higher catalytic activity than other materials with a hydrogen formation rate of 834 mL.min-1g-1 at room temperature and an activation energy of 31.6 KJ/mol.57
Metal-free systems as catalysts in dehydrogenation of AB
Some studies have reported using metal-free systems, specifically acids, as catalysts in the dehydrogenation of AB because all the previously discussed catalysts are transition metals which can be expensive or lead to environmental impacts.58–60 Though the efficacy of metal free catalysts is limited in H2 evolution of AB, Baker and co-workers investigated the acid catalysed hydrogen elimination of AB using [B(C6F5), HOSO2CF3] as catalysts. In this study 1.3 equivalents H2 per AB molecule was reported to be released by a 0.5 mol% catalyst loading.61 Although these fluorinated acids are toxic, corrosive and persistent in the environment, they have been reported to be effective catalysts for ammonia borane dehydrogenation. Lu et al. developed a metal-free bis(borane) Lewis acid catalyst to promote the H2 evolution of up to 2.5 equivalents per AB molecule. The dehydrogenation can be controlled on demand by heating to a moderate temperature (60℃) or cooling to room temperature. It was also noted that the catalyst can be reused multiple times without loss of activity.30 Nguyen et al. demonstrated that the Lewis acid BH3, which is generated from the decomposition of the B-N bond in AB, can act as an active bifunctional catalyst for favourable thermal dehydrogenation of AB. It is reported that the reaction energy barrier for H2 elimination of AB is reduced from 152.3 kJ/mol to 25.1 kJ/mol in the presence of BH3 catalyst resulting in AB compounds being touted as potential dihydrogen sources for hydrogen storage applications.7
Improved hydrogen elimination in AB derivatives with catalysts
Derivatives of AB with low molecular weight and high hydrogen content can be broadly used as chemical hydrogen storage materials. Hydrogen production from AB derivatives has been widely investigated by methods such as catalytic hydrolysis/thermolysis dehydrogenation, methanolysis and catalytic dehydrocoupling. The effective catalysts that can be applied for hydrolysis and/or thermolysis or dehydrocoupling of the derivatives of AB will be discussed in this section.
Dehydrogenation of methylamine borane with catalysts
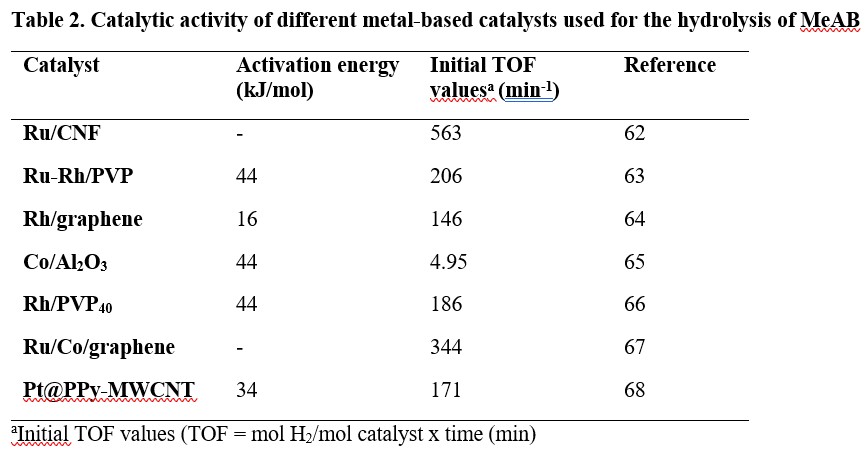
For the hydrolytic dehydrogenation of methylamine borane (MeAB), various heterogenous catalysts such as Ru supported on carbon nanofibers (Ru/CNF),62 polyvinylpyrrolidone (or polyvidone: PVP) stabilised Ru and Rh (Ru-Rh/PVP),63 Rh/graphene,64 Co/Al2O3,65 polyvidone stabilised Rh (Rh/PVP40),66 Ru/Co/graphene67 and Pt nanoparticles supported on polypyrrole-multi walled carbon nanotube (Pt@PPy-MWCNT)68 have been reported. Their activation energies and turnover frequencies (TOF) towards hydrolysis of MeAB are summarised in Table 2.
According to the literature, ruthenium-based nanomaterials show significant catalytic activity in dehydrogenation of MeAB during hydrolysis among other catalysts.62 Recently, ruthenium nanoparticles supported on sulfonated reduced graphene oxide (Ru@rGO-SO3H) have been synthesised and proved an effective catalyst in the hydrolytic hydrogen elimination of MeAB under mild conditions. Ru@rGO-SO3H nanocatalyst gives a high catalytic performance (TOF = 88 min-1) and near perfect conversion (> 99%) in the hydrolytic hydrogen formation from MeAB at 25℃. It is worth noting that this catalyst is very stable and is also reusable (> 95% after the 5th reuse).69
Dehydrogenation of dimethylamine borane with catalysts
Dimethylamine borane (DMAB) is another AB derivative which has an acceptable hydrogen content. The use of a suitable catalyst makes the dehydrogenation of DMAB easy at room temperature. Although various homogeneous and heterogeneous catalysts involving palladium, ruthenium, platinum and rhodium have been investigated,70–72 Pd-based nanomaterials73,74 demonstrated higher stability and the catalytic activity for the dehydrogenation or dehydrocoupling reactions of DMAB. Use of the graphene oxide stabilised bimetallic PdCo nanoparticles for dehydrocoupling of DMAB exhibited a high catalytic activity with a TOF of 3.78 min-1. Kinetic investigation revealed that a very high negative entropy (-170.85 Jmol-1K-1) and a low activation energy (17.53 ± 2 kJmol-1) resulted in the dehydrocoupling of DMAB by PdCo@GO catalyst.75 These values indicate that there is an associative mechanism in the transition state of the reaction facilitating the dehydrocoupling.
Dehydrogenation of hydrazine borane with catalysts
Hydrazine borane (HB) is one of the most hydrogen-dense derivatives of AB. Hydrogen present in HB can be eliminated with a suitable catalyst through hydrolysis of the BH3 group as well as decomposition of N2H4 moiety. To maximise the efficiency of HB as a promising hydrogen storage material, hydrolysis of the BH3 group of HB should only involve hydrogen production from HB rather than decomposition of the N2H4 group as well which would evolve hazardous ammonia gas alongside hydrogen. Monometallic catalysts such as Cu, Pd, Ru and Ni are reported in this context and only hydrolyse the BH3 of HB to generate H276–79 resulting in low hydrogen storage capacity.
Karahan et al. tested rhodium (III) chloride salt and obtained a TOF value of 200 min-1 for the hydrolysis of HB to release approximately 3.0 equivalents of H2 at room temperature.80 Another study of hydrolysis of HB was reported by Demirci and Xu, employing Ni0.89Pt0.11 nanomaterial as the catalyst. They demonstrated that 5.79 equivalents of H2 per HB can be released by this NiPt catalyst.81 Yao et al. reported the use of Ni0.6Pd0.4 nanoparticles modified with MoOx as a catalyst providing TOF values of 6.75 min-1 and almost 100% hydrogen selectivity at 50 °C.82 Recently, poly(N-vinyl-2-pyrrolidine)-stabilized CoRu nanoclusters have been synthesised to catalyse hydrolysis of HB and demonstrated a TOF value of 90 min-1.83
Dehydrogenation of hydrazine bisborane with catalysts
Dehydrogenation of HBB can be accelerated using a metal-based catalyst but the bimetallic NiPt catalyst is more active and can also decompose the N2H4 moiety. The Ni0.7Pt0.3 system was observed to be capable of eliminating H2 from HBB at 30℃ with the best kinetics by the hydrolysis of BH3 groups and was also able to decompose the N2H4 moiety at 70℃ with lower kinetics. However, dehydrogenation of HBB was not complete and reached only 80% conversion.84
Self-catalysis of cyclic ABs
The catalytic effect of BH3 on the H2 evolution reaction of various cyclic amine boranes (CABs) has been investigated. In the case of aziridine borane the activation energy of the dehydrogenation reaction is reduced from 163.2 to 84.4 kJ/mol in the presence of a BH3 catalyst.22 The BH3 catalysed dehydrogenation reaction of azetidine borane has a lower barrier than the predicted B-N bond dissociation energy thus favoring hydrogen elimination of azetidine borane over dissociation of the adduct.26 A theoretical interpretation of the catalytic role of BH3 on dehydrogenation from CAB adducts CnH2n+1N.BH3 (n = 2 – 5) showed that the dehydrogenation barrier height is reduced by 129.3, 133.9, 134.3 and 131.0 kJ/mol for aziridine, azetidine, pyrrolidine and piperidine borane respectively.25 The BH3-catalysed and BH3-uncatalysed hydrogen release pathway from pyrrolidine and piperidine borane have also been explored, where the BH3 catalyst was found to decrease the barrier height for the dehydrogenation reaction by around 129.7 and 133.9 kJ/mol respectively. As a result, BH3-catalysed hydrogen elimination has been determined to be a favorable process for both of these complexes.85
Conclusions
The catalytic role in enhanced dehydrogenation of linear and cyclic amine boranes has been reviewed. Although precious metals such as Rh, Ru, Pt and Ir exhibit much higher catalytic activities compared to the first-row transition metals Ni and Co in the hydrolysis of ABs and AB derivatives, their practical applications are restricted due to high cost and limited resources. The use of bimetallic nanoparticles containing a precious metal with a cost effective non-noble metal can significantly provide comparable catalytic activity in AB hydrolysis and H2 generation.
Further, transition metal nanoparticles supported by various stabilisers such as carbon black, metal oxides, MOFs and zeolites have attracted considerable interest to make the AB adduct an efficient H2 reservoir. These stabilising agents can prevent aggregation of metallic nanoparticles and result in enhanced catalytic activity and increased surface area. A metal-free catalyst (BH3 which is formed by cleavage of the B-N bond in AB) plays a significant catalytic role in decreasing the reaction barrier for the dehydrogenation reaction of both linear and various cyclic ABs. However, its application is limited because of the lower efficiency.
Future work will ideally focus on innovation around the use of an efficient catalyst to promote hydrogen elimination from AB and its derivatives to greatly enhance the use of chemical hydrogen storage systems whilst mitigating known limitations.
References
1. Wang, P.; Kang, X. Dalton Trans. 2008, 40, 5400–5413.
2. Jiang, H. L.; Singh, S. K.; Yan, J. M.; Zhang, X. B.; Xu, Q. ChemSusChem. 2010, 541–549.
3. Ramachandran, P. V.; Gagare, P. D. Inorg. Chem. 2007, 46 (19), 7810–7817.
4. Sanyal, U.; Demirci, U. B.; Jagirdar, B. R.; Miele, P. ChemSusChem. 2011, 1731–1739.
5. Kim, S. K.; Han, W. S.; Kim, T. J.; Kim, T. Y.; Nam, S. W.; Mitoraj, M.; Piekoś, L.; Michalak, A.; Hwang, S. J.; Kang, S. O. J. Am. Chem. Soc. 2010, 132 (29), 9954–9955.
6. Jaska, C. A.; Temple, K.; Lough, A. J.; Manners, I. J. Am. Chem. Soc. 2003, 125 (31), 9424-9434.
7. Nguyen, M. T.; Nguyen, V. S.; Matus, M. H.; Gopakumar, G.; Dixon, D. A. J. Phys. Chem. A 2007, 111 (4), 679–690.
8. Chandra, M.; Xu, Q. J. Power Sources 2006, 156 (2), 190–194.
9. Wu, H.; Cheng, Y.; Fan, Y.; Lu, X.; Li, L.; Liu, B.; Li, B.; Lu, S. Int. J. Hydrog. Energy 2020, 30325–30340.
10. Mal, S. S.; Stephens, F. H.; Baker, R. T. Chem. Commun. 2011, 47 (10), 2922–2924.
11. Hügle, T.; Kühnel, M. F.; Lentz, D. J. Am. Chem. Soc. 2009, 131 (21), 7444–7446.
12. Neiner, D.; Karkamkar, A.; Bowden, M.; Joon Choi, Y.; Luedtke, A.; Holladay, J.; Fisher, A.; Szymczak, N.; Autrey, T. Energy Environ. Sci. 2011, 4 (10), 4187–4193.
13. Groshens, T. J.; Hollins, R. A. Chem. Commun. 2009, 21, 3089–3091.
14. Zhu, Q. L.; Xu, Q. Energy Environ. Sci. 2015, 478–512.
15. Zhu, Q. L.; Zhong, D. C.; Demirci, U. B.; Xu, Q. ACS Catal. 2014, 4 (12), 4261–4268.
16. Goubeau, J.; Ricker, E. Z. Anorg. Allg. Chem 1961, 310, 123–142. https://doi.org/10.1002/zaac.19613100303
17. Moury, R.; Moussa, G.; Demirci, U. B.; Hannauer, J.; Bernard, S.; Petit, E.; Van Der Lee, A.; Miele, P. Phys. Chem. Chem. Phys. 2012, 14 (5), 1768–1777.
18. Yao, Q.; Lu, Z. H.; Wang, Y.; Chen, X.; Feng, G. J. Phys. Chem. C 2015, 119 (25), 14167–14174.
19. Şencanli, S.; Karahan, S.; Özkar, S. Int. J. Hydrog. Energy 2013, 38 (34), 14693–14703.
20. Özhava, D.; Kiliçaslan, N. Z.; Özkar, S. Appl. Catal. B 2015, 162, 573–582.
21. Sun, W.; Gu, Q.; Guo, Y.; Guo, Z.; Liu, H.; Yu, X. Int. J. Hydrog. Energy 2011, 36 (21), 13640–13644.
22. Németh, B.; Khater, B.; Guillemin, J. C.; Veszprémi, T. Inorg. Chem. 2010, 49 (11), 4854–4864.
23. Németh, B.; Guégan, J. P.; Veszprémi, T.; Guillemin, J. C. Inorg. Chem. 2013, 52 (1), 346–354.
24. Banu, T.; Sen, K.; Ghosh, D.; Debnath, T.; Das, A. K. RSC Adv. 2014, 4 (3), 1352–1361.
25. Sen, K.; Banu, T.; Debnath, T.; Ghosh, D.; Das, A. K. RSC Adv. 2014, 4 (42), 21924–21938.
26. Ja’O, A. M.; Masters, S. L.; Wann, D. A.; Rankine, C. D.; Nunes, J. P. F.; Guillemin, J. C. J. Phys. Chem. A 2019, 123 (32), 7104–7112.
27. Denney, M. C.; Pons, V.; Hebden, T. J.; Heinekey, D. M.; Goldberg, K. I. J. Am. Chem. Soc. 2006, 128 (37), 12048–12049.
28. Blaquiere, N.; Diallo-Garcia, S.; Gorelsky, S. I.; Black, D. A.; Fagnou, K. J. Am. Chem. Soc. 2008, 130 (43), 14034–14035.
29. Kim, S. K.; Han, W. S.; Kim, T. J.; Kim, T. Y.; Nam, S. W.; Mitoraj, M.; Piekoś, L.; Michalak, A.; Hwang, S. J.; Kang, S. O. J. Am. Chem. Soc. 2010, 132 (29), 9954–9955.
30. Lu, Z.; Schweighauser, L.; Hausmann, H.; Wegner, H. A. Angew. Chem. 2015, 127 (51), 15777–15780.
31. Li, P. Z.; Aijaz, A.; Xu, Q. Angew. Chem. Int. Ed. 2012, 51 (27), 6753–6756.
32. Yang, X.; Cheng, F.; Tao, Z.; Chen, J. J. Power Sources 2011, 196 (5), 2785–2789.
33. Liu, M.; Zhou, L.; Luo, X.; Wan, C.; Xu, L. Catalysts 2020, 1–34.
34. Rakap, M. Appl. Catal. B 2015, 163, 129–134.
35. Li, S. J.; Wang, H. L.; Yan, J. M.; Jiang, Q. Int. J. Hydrog. Energy 2017, 42 (40), 25251–25257.
36. Din, M.; Metin, Ö.; Özkar, S. Catal. Today 2012, 183 (1), 10–16.
37. Wang, Q.; Fu, F.; Yang, S.; Martinez Moro, M.; Ramirez, M. D. L. A.; Moya, S.; Salmon, L.; Ruiz, J.; Astruc, D. ACS Catal. 2019, 9 (2), 1110–1119.
38. Zahmakiran, M.; Özkar, S. Top. Catal. 2013, 56 (13–14), 1171–1183.
39. Regalbuto, J. Catalyst Prepartion: Science and Engineering; BocaRaton: Taylor & Francis, 2007.
40. Akbayrak, S.; Özkar, S. ACS Appl. Mater. Interfaces 2012, 4 (11), 6302–6310.
41. Du, C.; Ao, Q.; Cao, N.; Yang, L.; Luo, W.; Cheng, G. Int. J. Hydrog. Energy 2015, 40 (18), 6180–6187.
42. Liang, H.; Chen, G.; Desinan, S.; Rosei, R.; Rosei, F.; Ma, D. Int. J. Hydrog. Energy 2012, 37 (23), 17921–17927.
43. Akbayrak, S.; Tonbul, Y.; Özkar, S. Dalton Trans. 2016, 45 (27), 10969–10978.
44. Yao, Q.; Lu, Z. H.; Yang, K.; Chen, X.; Zhu, M. Sci. Rep. 2015, 5.
45. Yang, K.; Zhou, L.; Yu, G.; Xiong, X.; Ye, M.; Li, Y.; Lu, D.; Pan, Y.; Chen, M.; Zhang, L.; Gao, D.; Wang, Z.; Liu, H.; Xia, Q. Int. J. Hydrog. Energy 2016, 41 (15), 6300–6309.
46. Akbayrak, S.; Tanyildizi, S.; Morkan, I.; Özkar, S. Int. J. Hydrog. Energy 2014, 39 (18), 9628–9637.
47. Wen, L.; Su, J.; Wu, X.; Cai, P.; Luo, W.; Cheng, G. Int. J. Hydrog. Energy 2014, 39 (30), 17129–17135.
48. Fan, G.; Liu, Q.; Tang, D.; Li, X.; Bi, J.; Gao, D. Int. J. Hydrog. Energy 2016, 41 (3), 1542–1549.
49. Tonbul, Y.; Akbayrak, S.; Özkar, S. J. Colloid Interface Sci. 2018, 513, 287–294.
50. Can, H.; Metin, Ö. Appl. Catal B 2012, 125, 304–310.
51. Song, P.; Li, Y.; Li, W.; He, B.; Yang, J.; Li, X. Int. J. Hydrog. Energy 2011, 36 (17), 10468–10473.
52. Liu, C. H.; Wu, Y. C.; Chou, C. C.; Chen, B. H.; Hsueh, C. L.; Ku, J. R.; Tsau, F. Int. J .Hydrog. Energy 2012, 37, 2950–2959.
53. Akdim, O.; Demirci, U. B.; Miele, P. Int. J. Hydrog. Energy 2011, 36 (21), 13669–13675.
54. Yan, J. M.; Zhang, X. B.; Shioyama, H.; Xu, Q. J. Power Sources 2010, 195 (4), 1091–1094.
55. Demirci, U. B.; Miele, P. J. Power Sources 2010, 195 (13), 4030–4035.
56. Sun, D.; Mazumder, V.; Metin, Ö.; Sun, S. ACS Nano. 2011, 5 (8), 6458–6464.
57. Zhou, L.; Zhang, T.; Tao, Z.; Chen, J. Nano. Res. 2014, 7 (5), 774–781.
58. Keaton, R. J.; Blacquiere, J. M.; Baker, R. T. J. Am. Chem. Soc. 2007, 129 (7), 1844–1845.
59. Jaska, C. A.; Temple, K.; Lough, A. J.; Manners, I. J. Am. Chem. Soc. 2003, 125 (31), 9424–9434.
60. Conley, B. L.; Guess, D.; Williams, T. J. J. Am. Chem. Soc. 2011, 133 (36), 14212–14215.
61. Stephens, F. H.; Baker, R. T.; Matus, M. H.; Grant, D. J.; Dixon, D. A. Angew. Chem. Int. Ed. 2007, 46 (5), 746–749.
62. Khalily, M. A.; Yurderi, M.; Haider, A.; Bulut, A.; Patil, B.; Zahmakiran, M.; Uyar, T. ACS Appl. Mater. Interfaces 2018, 10 (31), 26162–26169.
63. Taçyıldız, S.; Demirkan, B.; Karataş, Y.; Gulcan, M.; Sen, F. J. Mol. Liq. 2019, 285, 1–8.
64. Shen, J.; Yang, L.; Hu, K.; Luo, W.; Cheng, G. Int. J. Hydrog. Energy 2015, 40 (2), 1062–1070.
65. Baguc, I. B.; Yurderi, M.; Bulut, A.; Celebi, M.; Kanberoglu, G. S.; Zahmakiran, M.; Kaya, M.; Aydemir, M.; Durap, F.; Baysal, A. Int. J. Hydrog. Energy 2019, 44 (53), 28441–28450.
66. Gulcan, M.; Karataş, Y. New J. Chem. 2017, 41 (20), 11839–11845.
67. Cao, N.; Su, J.; Luo, W.; Cheng, G. Catal. Commun. 2014, 43, 47–51.
68. Gulcan, M.; Karataş, Y. New J. Chem. 2017, 41 (20), 11839–11845.
69. Yıldırım, R.; Gülcan, M. Int. J. Hydrog. Energy 2021, 46 (64), 32523–32535.
70. Friedrich, A.; Drees, M.; Schneider, S. Chem. Eur. J. 2009, 15 (40), 10339–10342.
71. Sloan, M. E.; Clark, T. J.; Manners, I. Inorg. Chem. 2009, 48 (6), 2429–2435.
72. Sen, B.; Kuzu, S.; Demir, E.; Akocak, S.; Sen, F. Int. J. Hydrog. Energy 2017, 42 (36), 23284–23291.
73. Çelik, B.; Yildiz, Y.; Sert, H.; Erken, E.; Koşkun, Y.; Şen, F. RSC Advances, 2016, 24097–24102.
74. Sen, B.; Kuzu, S.; Demir, E.; Yıldırır, E.; Sen, F. Int. J. Hydrog. Energy 2017, 42 (36), 23307–23314.
75. Şen, B.; Aygün, A.; Şavk, A.; Çalımlı, M. H.; Gülbay, S. K.; Şen, F. Int. J. Hydrog. Energy 2020, 45 (5), 3569–3576.
76. Yao, Q.; Lu, Z. H.; Wang, Y.; Chen, X.; Feng, G. J. Phys. Chem. C 2015, 119 (25), 14167–14174.
77. Tunç, N.; Abay, B.; Rakap, M. J. Power Sources 2015, 299, 403–407.
78. Yao, Q.; Lu, Z. H.; Yang, K.; Chen, X.; Zhu, M. Sci. Rep. 2015, 5.
79. Şencanli, S.; Karahan, S.; Özkar, S. Int. J. Hydrog. Energy 2013, 38 (34), 14693–14703.
80. Karahan, S.; Zahmakran, M.; Özkar, S. Int. J. Hydrog. Energy 2011, 36 (8), 4958–4966.
81. Hannauer, J.; Akdim, O.; Demirci, U. B.; Geantet, C.; Herrmann, J. M.; Miele, P.; Xu, Q. Energy Environ. Sci. 2011, 4 (9), 3355–3358.
82. Yao, Q.; Yang, K.; Nie, W.; Li, Y.; Lu, Z. H. Renew. Energy 2020, 147, 2024–2031.
83. Rakap, M. Int. J. Hydrog. Energy 2020, 45 (31), 15611–15617.
84. Petit, E.; Demirci, U. B. Int. J. Hydrogen Energy 2018, 43 (3), 1261–1270.
85. Ja’o, A. M.; Wann, D. A.; Rankine, C. D.; Nunes, J. P. F.; Guillemin, J. C.; Masters, S. L. Struct. Chem. 2021, 32 (1), 205–213.