Introduction
Oligonucleotide synthesis is a method of producing short single-stranded DNA and RNA sequences using chemistry, rather than obtaining it from biological samples. Multiple methods exist for DNA extraction from biological samples, but the outputs of these methods often include a wide range of sequences which can be difficult to isolate. In comparison, methods for chemical DNA synthesis have the advantage of producing only the desired sequence of nucleotides and obtaining DNA with modifications and labels for various applications. Over several decades, the automated synthesis of short DNA and RNA molecules using nucleoside phosphoramidites (Fig. 1A) has been widely used and applied in various scientific fields ranging from material science to gene therapy and biomanufacturing. For example, DNA primers produced using nucleoside phosphoramidite chemistry were recently used to amplify viral RNA in COVID-19 tests. However, this method is mainly restricted by the length of DNA and RNA molecule produced (200 nucleotides).1 Recently, Ansa Biotechnologies, Inc., a biotech company located in the USA, have reported new enzymatic DNA synthesis which enable similarly controllable synthesis of oligonucleotides of considerably longer lengths.2,3
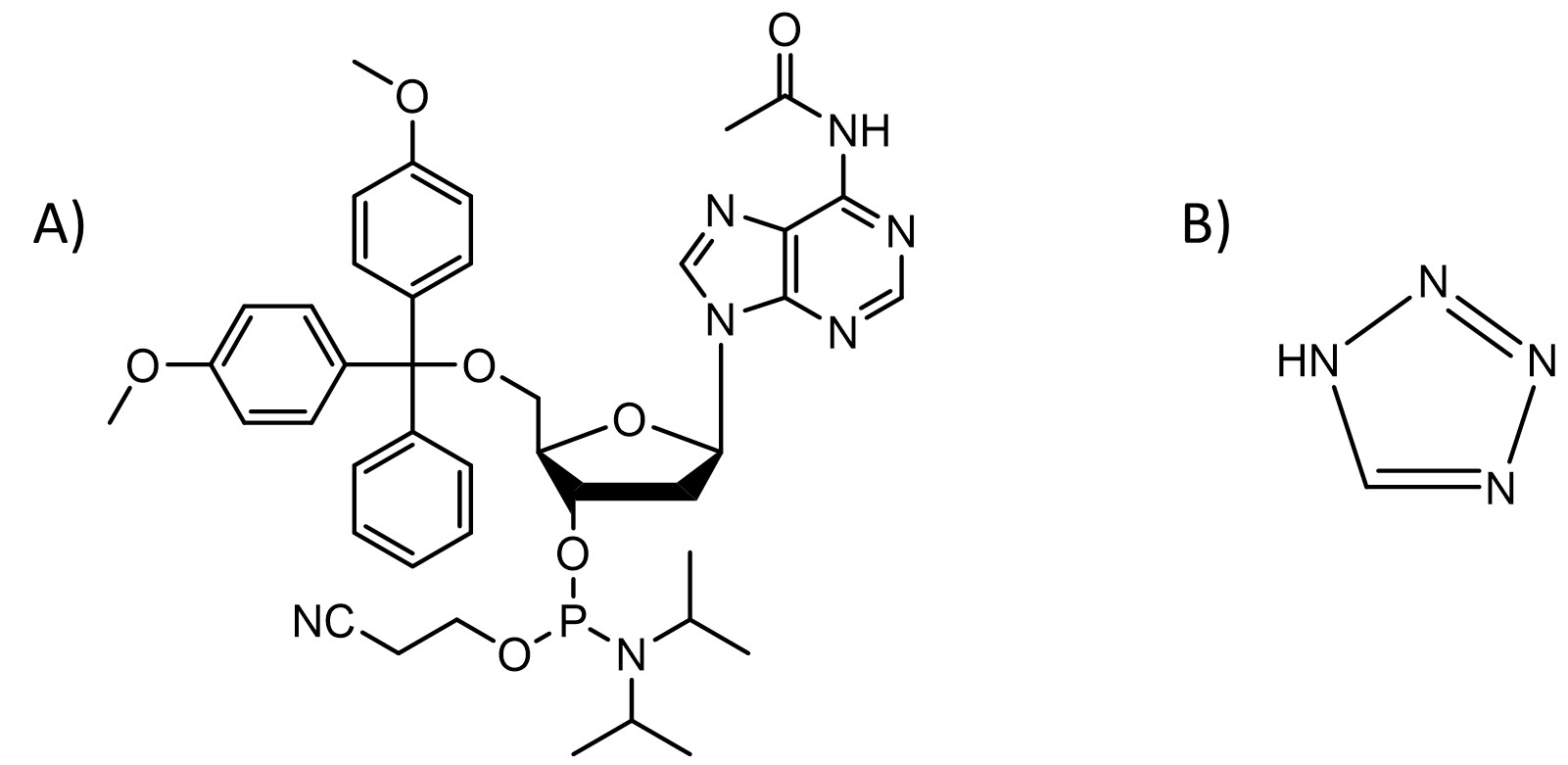
Classical DNA and RNA synthesis
Nucleoside phosphoramidites are the most common chemicals used for producing short DNA and RNA.1 This method is usually carried out as a solid-phase synthesis meaning the oligonucleotide is assembled on a glass with controlled pore sizes (500 - 2000 Å). The support is placed in a column between two filters and all solutions are passed through it enabling use of a large excess of reagents to push the reaction to completion and removal of byproducts and excess reagents by washing support with a suitable solvent. This process consists of the following four steps:
1. Deblocking: a 5′-OH protecting group, usually 4,4′-dimethoxytrityl (DMT), is cleaved from the oligonucleotide attached to the CPG in acidic conditions using trichloroacetic acid dissolved in dichloromethane (Fig. 2i).
2. Coupling: the deprotected 5′-OH is coupled with the 5′-DMT-3′-nucleoside phosphoramidite, such as the one shown in Fig. 1A, (10 - 40 equivalents) of the next base in the sequence using a slightly acidic activator such as 1H-tetrazole (100-250 equivalents, pKa = 4.75, Fig. 1B). All reagents are dissolved in dry acetonitrile. This most critical step requires anhydrous conditions as any water present in the solvent or reagents will outcompete a primary 5′-OH present on the support (Fig. 2ii).
3. Capping: unreacted 5′-OH groups are capped with acetyl protecting groups to prevent formation of sequences with missing bases (Fig. 2iii).
4. Oxidation: the trivalent phosphorous is oxidised to a pentavalent phosphate using I2 in the presence of pyridine and water (Fig. 2iii).
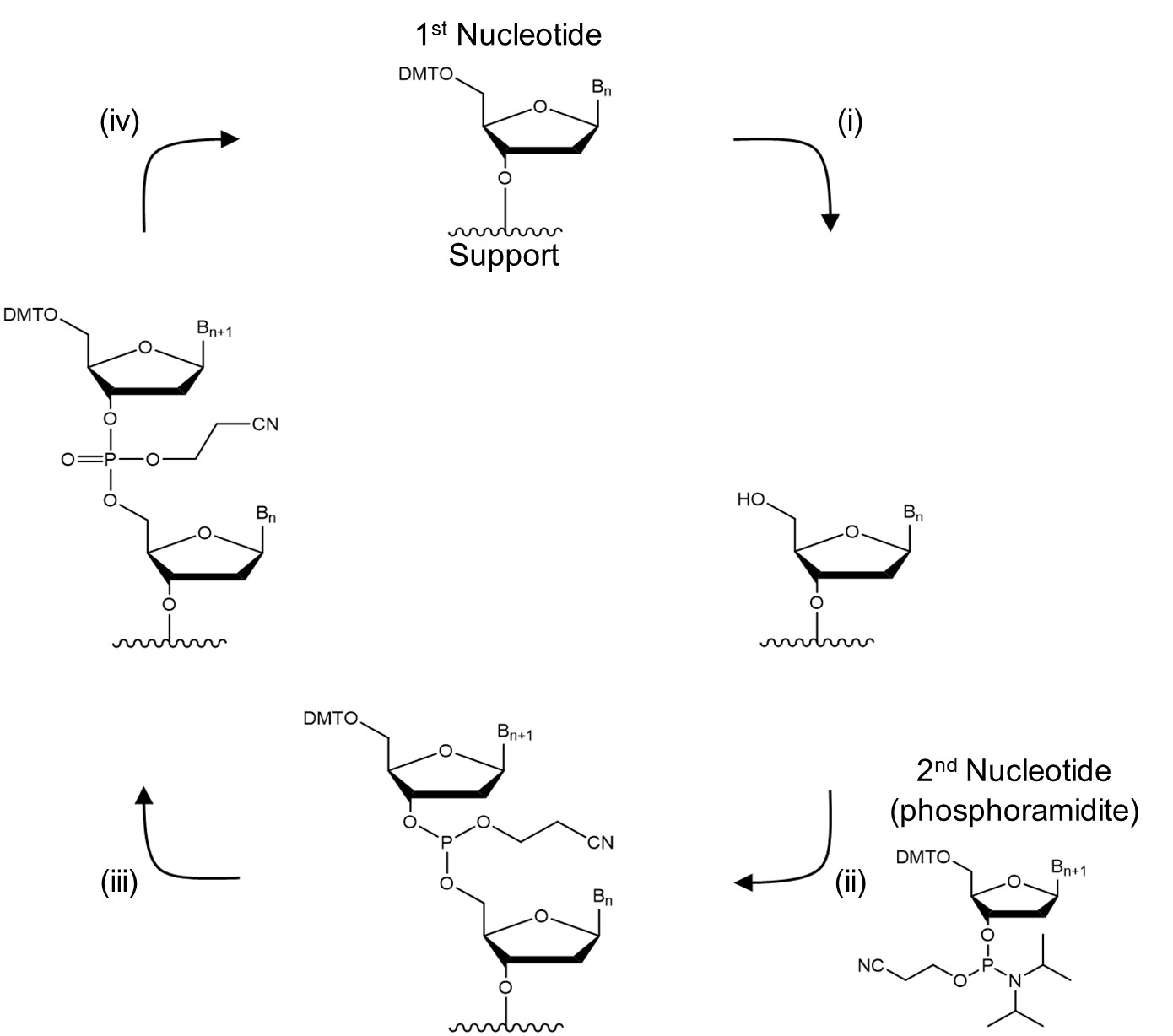
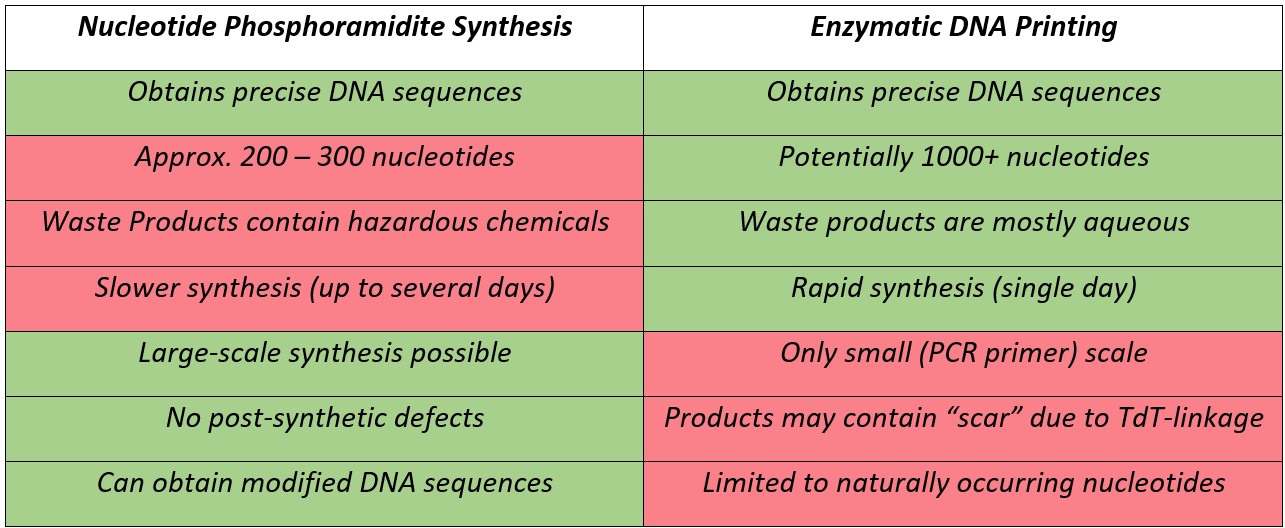
An eagle eye will notice that the synthesis occurs in the 3′ to 5′ direction which is opposite to the biological synthesis of DNA. This process can be repeated until the desired sequence is obtained, which is then removed from the solid support by cleaving the linkage between the first nucleotide and the support under basic conditions (Fig. 2iv). By adding a single base in each cycle and capping unreacted material the final sequence can be precisely controlled. However, the large number of steps means that each individual step must be highly efficient (preferably > 99% yield) and even so the length of sequences which can be obtained with reasonable yield is limited. 200 – 300 nucleotides is usually considered the upper limit of this technique.
Enzymatic DNA printing
In 2018, Palluk et al.2 reported the stepwise synthesis of a 10-mer oligonucleotide using terminal deoxynucleotidyl transferase (TdT), a polymerase enzyme which adds nucleotides to the 3′ terminus of DNA. They highlighted the need to find a suitable solid support and improve coupling efficiency to consistently be above 99%. After further development of this technique, Ansa Biotechnologies, Inc., a company based in San Francisco, USA, announced that they obtained a 1005 nucleotide sequence3, significantly longer than any sequence produced using nucleoside phosphoramidites. When the products of this technique were amplified using the polymerase chain reaction (PCR, a technique used to replicate existing DNA sequences), 28% of molecules contained the correct oligonucleotide sequence. The large number of steps involved in this sequence means that 28% corresponds to each step having an average yield upwards of 99.9% for each step. Their method (Fig. 3A) uses deoxyribonucleoside triphosphates (dNTPs), which are used for DNA synthesis in nature but here dNTPs are attached covalently to the TdT enzyme (Fig. 3B). Creating these conjugates required the use of modified nucleotides, which were converted to the intended nucleotide during the deprotection step. The TdT-dNTP conjugate forms a complex with a single stranded DNA sequence immobilised on a solid support and the dNTP is coupled to the free 3′-OH, as shown in Fig. 3i. The linkage between TdT and the oligonucleotide is then cleaved as shown in Fig. 3ii. The next TdT-dNTP conjugate can be added, allowing stepwise controllable addition of nucleotides, similar to nucleoside phosphoramidite synthesis. One downside of this method is the inclusion of a “scar” resulting from incomplete cleavage of the linker to TdT. The size of the scar has been reduced across multiple iterations of this method but is necessary to allow for this linker chemistry.
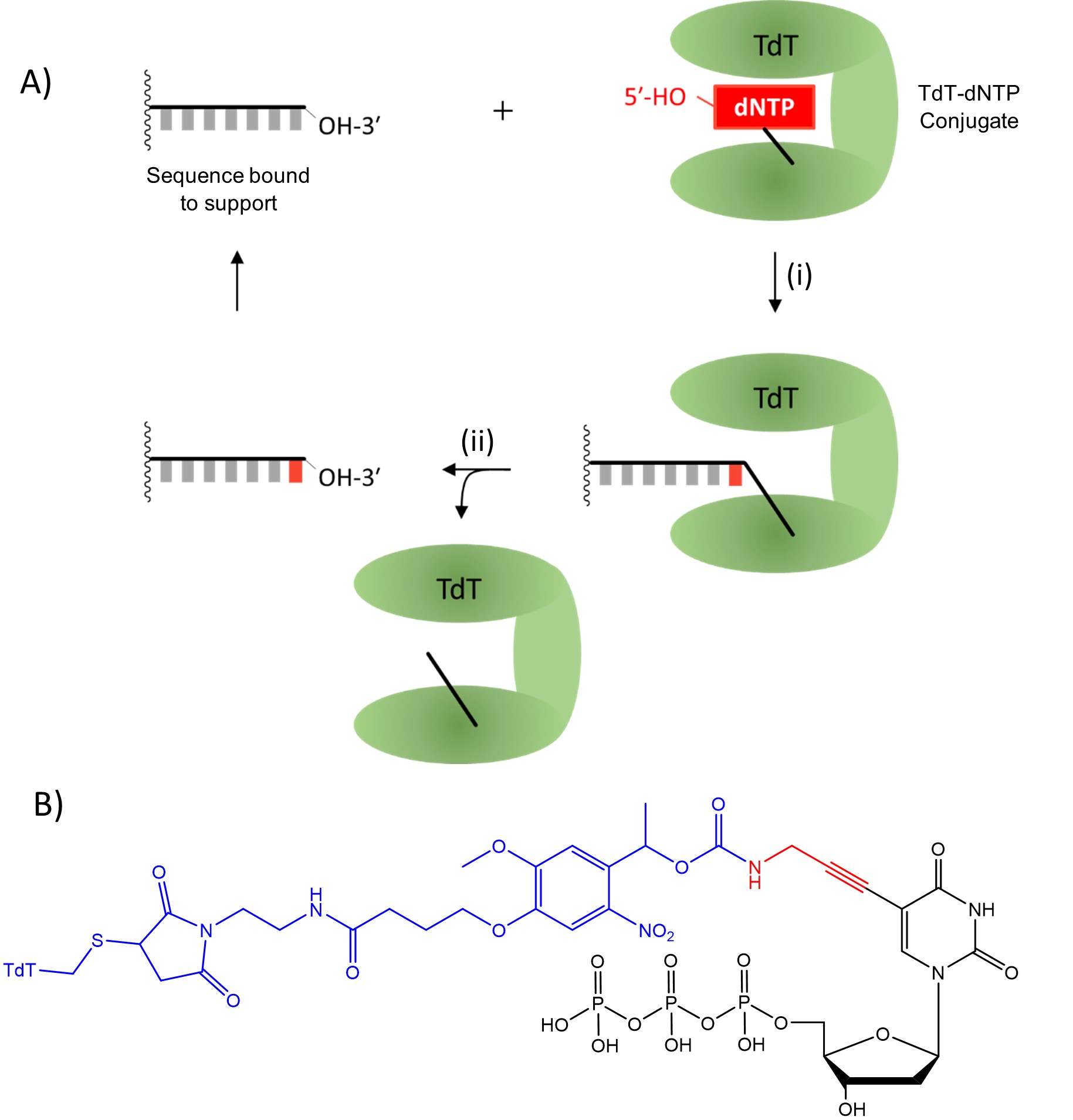
The use of TdT-dNTP conjugates retains the advantageous stepwise addition of nucleoside phosphoramidite chemistry, allowing for the precise synthesis of specific oligonucleotides which are otherwise difficult to study in isolation. However, it is also suggested that they offer significantly increased coupling efficiency compared to nucleoside phosphoramidites, allowing for much longer sequences to be synthesised and studied. This is possible both because the individual coupling steps are enzymatic and highly efficient and because each nucleotide addition cycle uses fewer steps than nucleoside phosphoramidite coupling, forgoing both oxidation and capping steps. The automation and commercialisation of this technique potentially gives researchers access to efficient synthesis of oligonucleotides with lengths up to one thousand nucleotides. Additionally, all reactions involved in TdT-dNTP coupling occur in aqueous conditions, significantly reducing the production of hazardous waste products. Sustainability is an increasing concern in chemistry and technology that utilises aqueous conditions while maintaining the efficiency of existing methods is becoming increasingly necessary. If TdT-dNTP conjugates can be produced at the scale required for DNA synthesis, then this development could represent a significant step forward for sustainable synthesis of oligonucleotides.
However, classical nucleoside phosphoramidite coupling retains several key advantages. The scale of TdT-dNTP based synthesis is not clear from their reports, but the requirement to use DNA amplification using PCR to scale up the products suggests that is lower than the scales possible with phosphoramidite chemistry. Production of PCR primers is one use of nucleoside phosphoramidite chemistry, and in that regard TdT-dNTP protocols may be advantageous, but the scalability of this protocol is uncertain at this stage. Furthermore, a wide range of modified phosphoramidites are commercially available due to the popularity of phosphoramidite coupling for DNA synthesis. Growing interest in TdT based coupling could result in a similar range of modified TdT conjugates, but these types of modifications would not allow for amplification with PCR, and all the modifications possible with phosphoramidite chemistry may not be translatable to TdT chemistry. One example is phosphate modifications carried out during the oxidation step of DNA synthesis, which cannot occur during TdT synthesis because there is no oxidation step.4-7 Additionally, as mentioned previously TdT synthesis leaves a “scar” as a result of the linker chemistry used, which does not occur in phosphoramidite synthesis. However, improvements in the linker chemistry since the original publication in 20182 could avoid this problem in future.
Conclusions
The newly reported development of enzymatic DNA printing using TdT-dNTP conjugates to synthesise oligonucleotides offers a highly efficient method of producing considerably longer oligonucleotides than standard nucleoside phosphoramidite synthesis, while retaining precise control of the DNA primary structure. Additionally, it avoids the production of numerous hazardous byproducts of phosphoramidite based coupling as all reactions take place in aqueous conditions. Ansa Biotechnologies, Inc. is beginning to offer a commercial source of nucleotides produced using this method, referred to as Ansamers, which provides a new method of creating and studying long chemically synthesised oligonucleotides.3 Similar technology is also available from DNA Script, Inc., also based in San Francisco, USA, which offers Hi-Fidelity kits intended to synthesise sequences up to 80 nucleotides also using TdT-based conjugates for synthesis. This technology appears to use different TdT-nucleotide linkers, which provides oligonucleotides without a “scar,” although precise details of the DNA Script synthetic technique do not appear to be publicly available. While this method offers several potential advantages compared to standard phosphoramidite chemistry (Table 1), it may lack the ability to scale and modify the oligonucleotides outputs which is possible with phosphoramidite chemistry, and therefore it remains to be seen if Ansamers are a viable alternative to the currently preferred method of DNA synthesis.
References
1. Caruthers, M. H. J. Biol. Chem. 2013, 288 (2), 1420-1427. DOI: 10.1074/jbc.X112.442855.
2. Palluk, S.; Arlow, D. H.; de Rond, T.; Barthel, S.; Kang, J. S.; Bector, R.; Baghdassarian, H. M.; Truong, A. N.; Kim, P. W.; Singh, A. K. et al. Nat. Biotechnol. 2018, 36 (7), 645-650. DOI: 10.1038/nbt.4173.
3. Ansa Biotechnologies Ltd. Ansa Biotechnologies Announces Successful de novo Synthesis of World’s Longest Oligonucleotide at 1005 Bases. 2023. https://www.businesswire.com/news/home/20230309005124/en/Ansa-Biotechnologies-Announces-Successful-de-novo-Synthesis-of-World%E2%80%99s-Longest-Oligonucleotide-at-1005-Bases (accessed 19/07/2023).
4. Kupryushkin, M. S.; Pyshnyi, D. V.; Stetsenko, D. A. Acta Naturae 2014, 6 (4), 116-118.
5. Chelobanov, B. P.; Burakova, E. A.; Prokhorova, D. V.; Fokina, A. A.; Stetsenko, D. A. Russian J. Bioorg. Chem. 2018, 43 (6), 664-668. DOI: 10.1134/s1068162017060024.
6. Kupryushkin, M. S.; Apukhtina, V. S.; Vasil’eva, S. V.; Pyshnyi, D. V.; Stetsenko, D. A. Izv. Akad. Nauk, Ser. Khim. 2015, 7, 1678-1681.
7. Su, Y.; Bayarjargal, M.; Hale, T. K.; Filichev, V. Bielstein J. Org. Chem. 2021, 17, 749-761.